Research
Our overarching goal is to use computational biology approaches to gain a better understanding of biological systems. One focus is to develop deep neural network architectures to analyse high-throughput cancer data and push forward the area of precision medicine. We are interested in integrative approaches that combine high-throughput data (genomics, transcriptomics, proteomics, metabolomics) on particular systems with existing biological knowledge from knowledge-bases such as biological network data (KEGG, Reactome). We are also investigating the use of multi-agent systems (MAS) to facilitate flexible and goal-driven analysis to integrate bioinformatics data and methods.
Here are some themes and techniques that we currently work on: Machine Learning and AI, Precision Medicine, Genomics / Genetics, Transcriptomics, Proteomics, Metabolomics, Systems Biology Integration
Machine Learning and AI
Bentham, Robert B., Kevin Bryson, and Gyorgy Szabadkai.
Nucleic Acids Research 45, no. 15 (6 September 2017): 8712–30.
MCbiclust is a novel method that can extract patterns from massive transcriptomics data collections. We show that it can effectively stratify patients based on their intrinsic gene expression signatures.
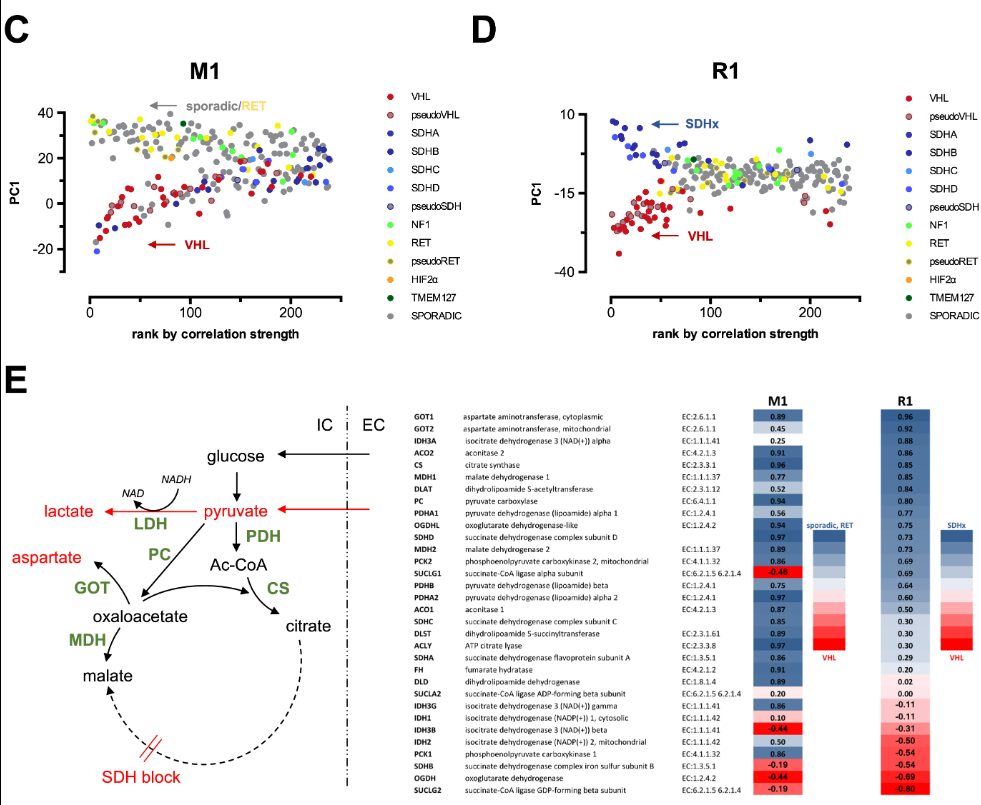
Bryson, K., M. Luck, M. Joy, and D. T. Jones.
Seminal paper outlining the GeneWeaver multi-agent system (MAS) for cooperative genome annotation.
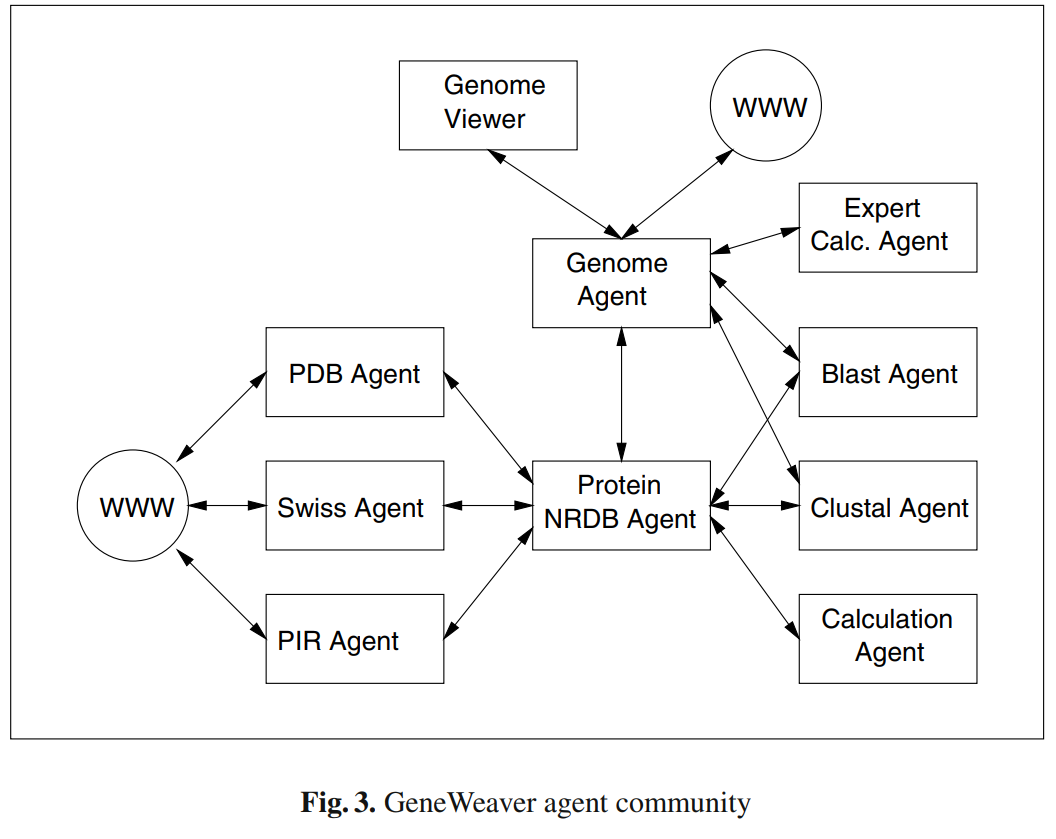
Malki, K., E. Koritskaya, F. Harris, K. Bryson, M. Herbster, and M. G. Tosto.
Translational Psychiatry 6, no. 6 (June 2016): e839–e839.
This applies machine learning to epigenetic data from identical twins where one has major depressive disorder and the other does not. This identifies biological networks that potentially involved in the diseased mechanism.
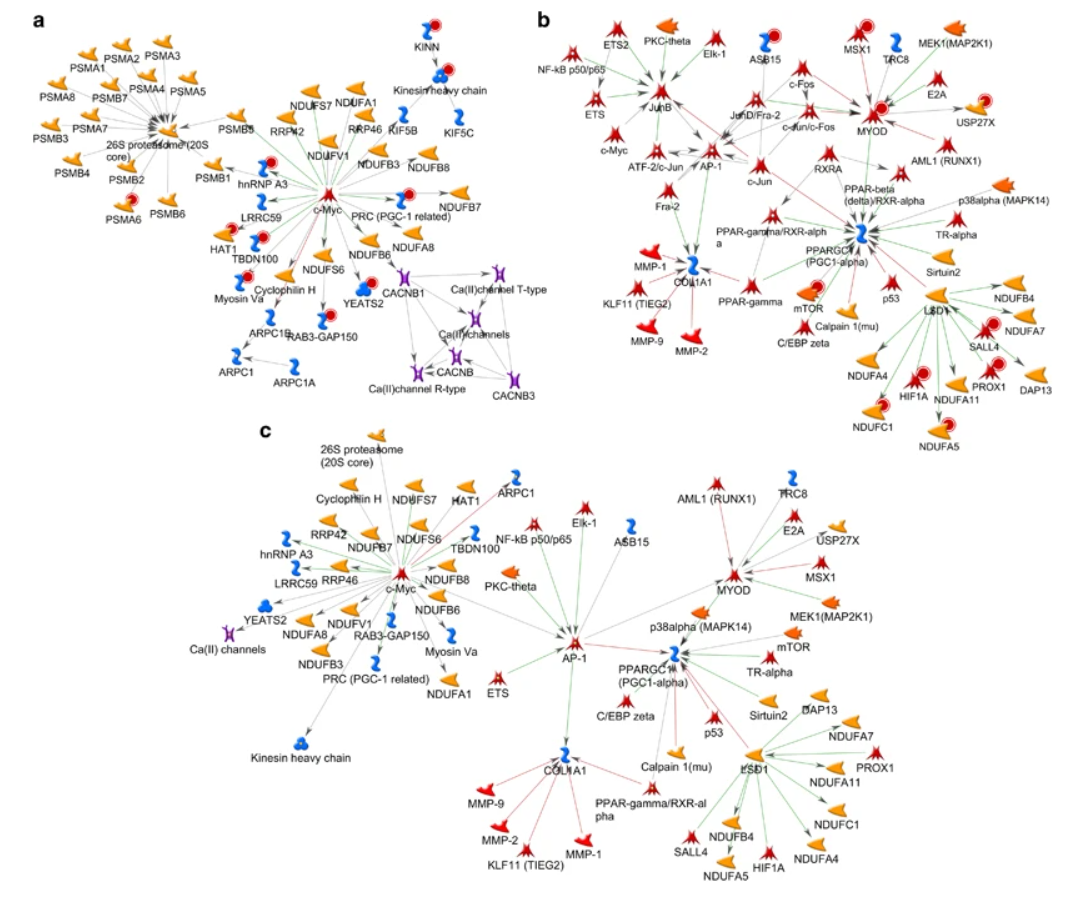
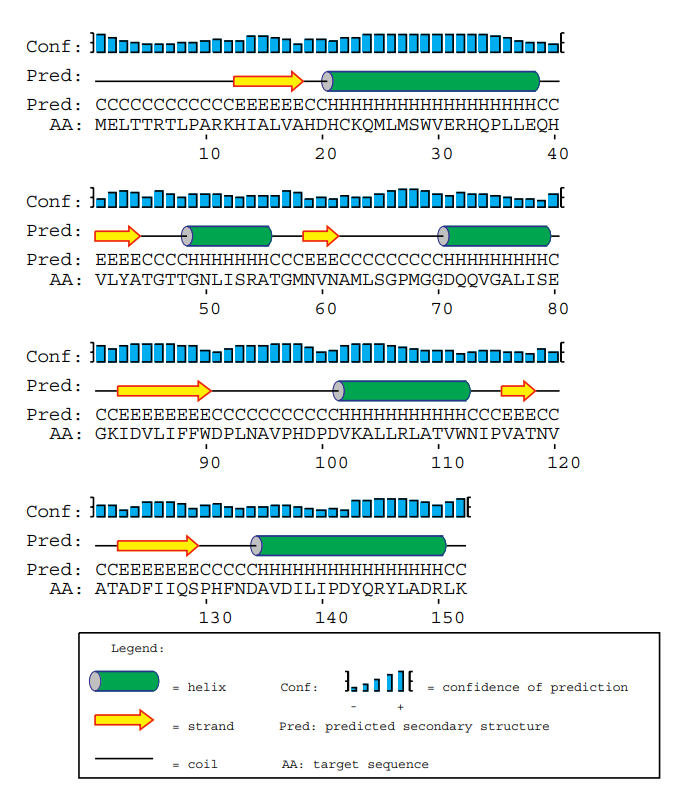
McGuffin, Liam J., Kevin Bryson, and David T. Jones.
Bioinformatics 16, no. 4 (1 April 2000): 404–5.
One of the most successful neural network architectures for predicting ab-initio secondary structure from protein sequences.
Medlar, Alan, Dorota Głowacka, Horia Stanescu, Kevin Bryson, and Robert Kleta.
Bioinformatics 29, no. 4 (15 February 2013): 413–19.
Human pedigree analysis is often used to identify regions of the genome that are invovled in rare diseases. The analysis of large human pedigrees is incredibly computational; particularly consanguineous ones which often results in rare recessive diseases showing themselves. In this paper we develop an parallel MCMC genetic linkage algorithm that uses multicore CPUs and GPUs, resulting in a magnitute improvement in speed over other popular methods at the time.
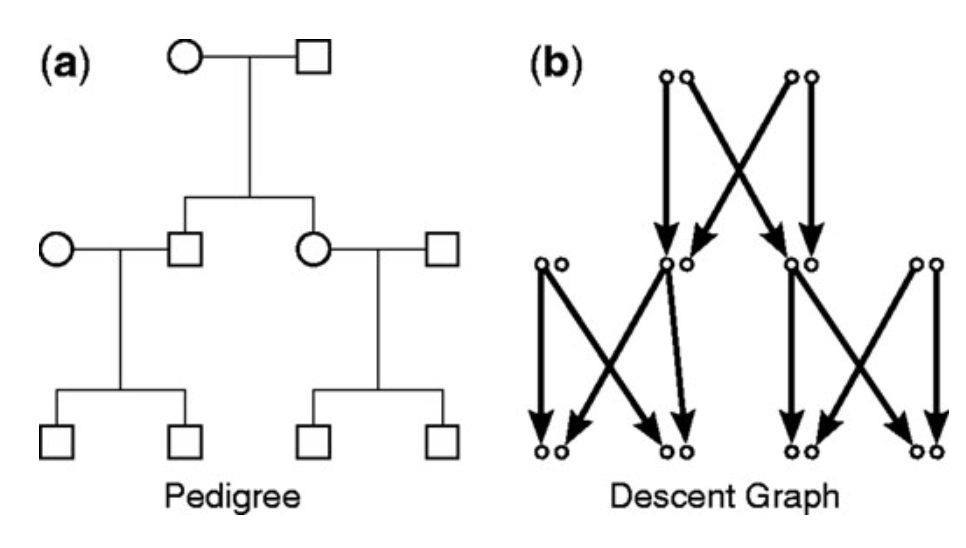
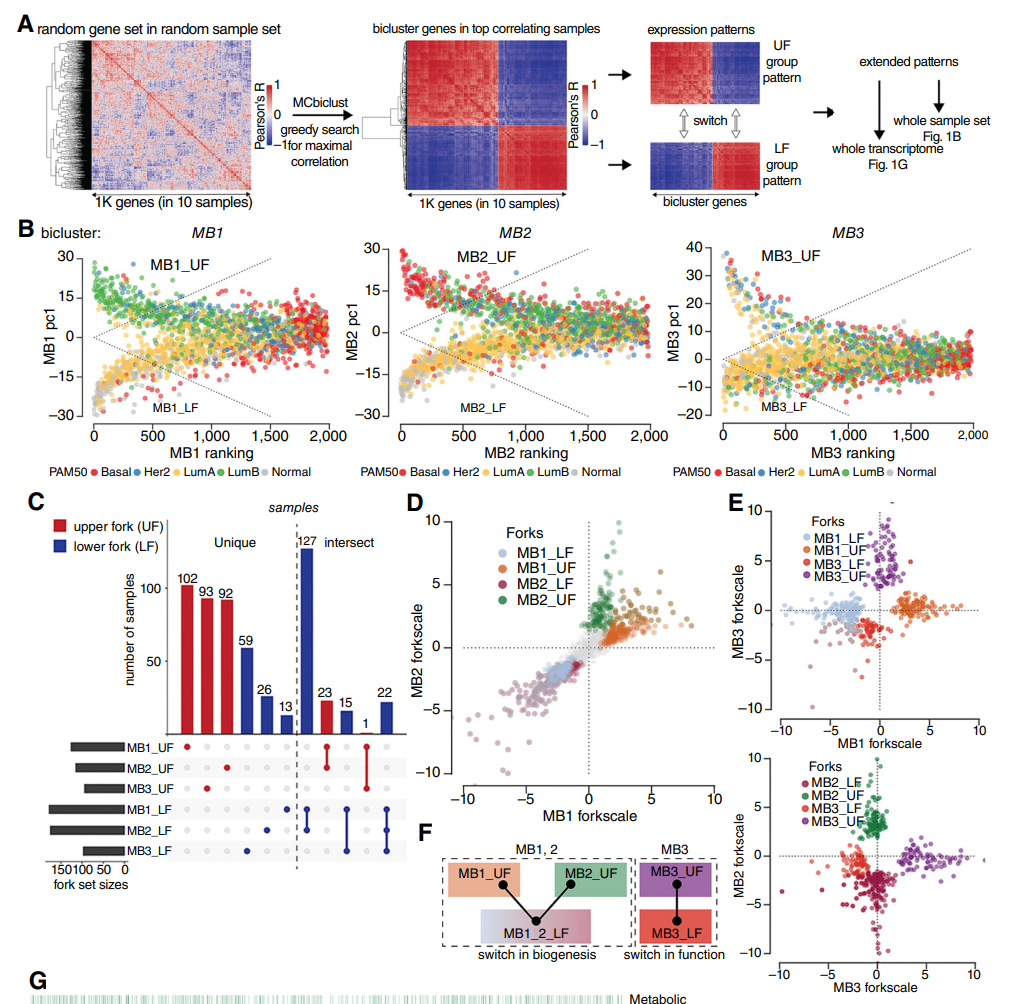
Menegollo, Michela, Robert B. Bentham, Tiago Henriques, Seow Q. Ng, Ziyu Ren, Clarinde Esculier, Sia Agarwal, et al.
Cancer Research 84, no. 17 (4 September 2024): 2911–25.
Application of our MCbiclust software to breast cancer samples and experimental validation using metabolomics.
Patel, Z, Patel, ZI, Robinson, J, Robertson, DP, Stein, JE, Chenery, P, Latham, K, Evseeva, I, Bryson, K, and Marsh, SGE.
25th British Society for Histocompatibility and Immunogenetics Conference
A new probabilistic matching algorithm was developed which was used by the Anthony Nolan Trust for bone marrow transplant matching.
Radivojac, Predrag, Wyatt T. Clark, Tal Ronnen Oron, Alexandra M. Schnoes, Tobias Wittkop, Artem Sokolov, Kiley Graim, et al.
Nature Methods 10, no. 3 (March 2013): 221–27.
A competition involving prediction Gene Ontology protein function prediction for protein sequences. The method of our research group at UCL was ranked first in the competition.
Viñas, Ramon, Helena Andrés-Terré, Pietro Liò, and Kevin Bryson.
Bioinformatics 38, no. 3 (12 January 2022): 730–37.
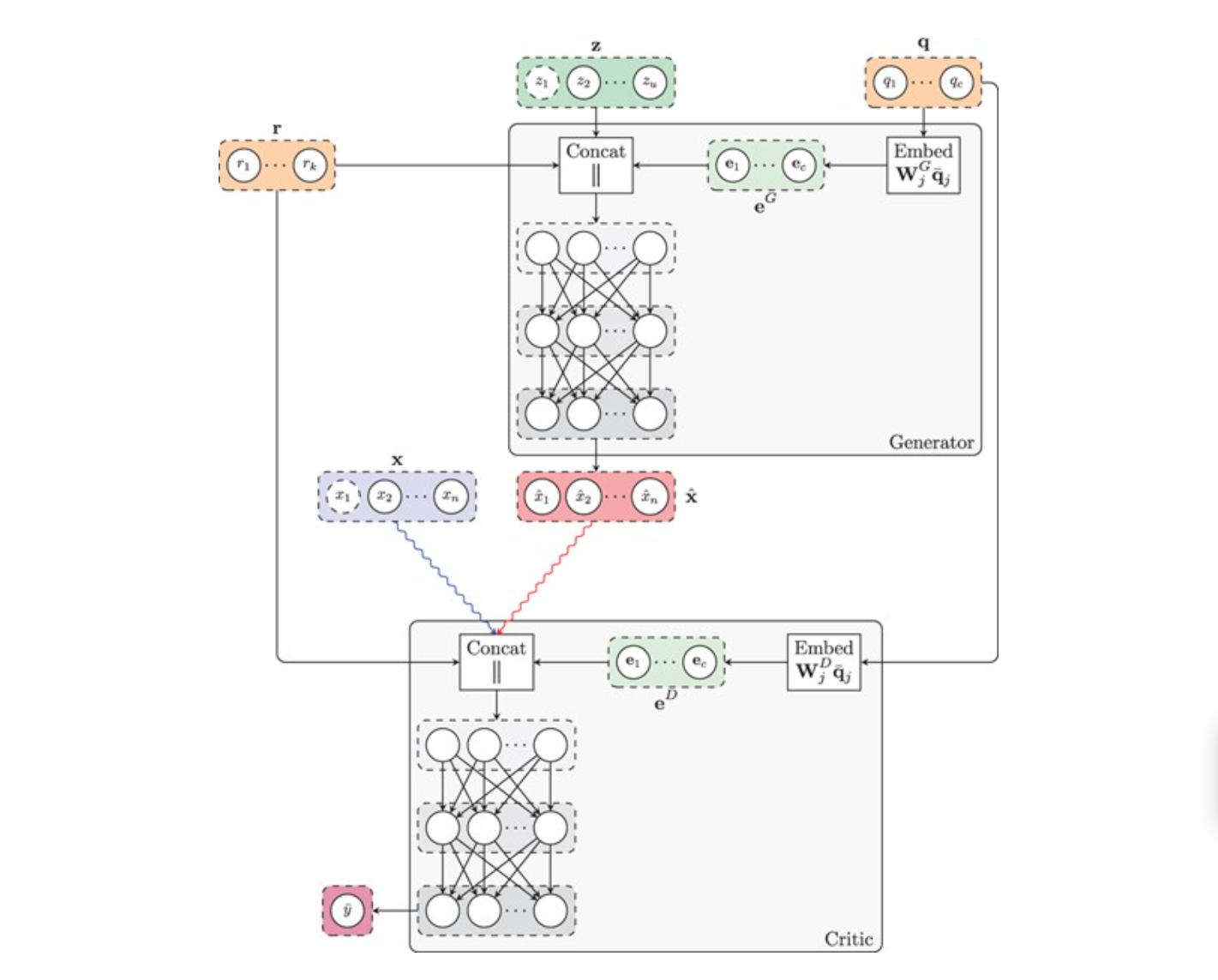
We introduce the first deep learning GAN method with word embeddings to simulate gene expression data for a wide range of biological systems from bacteria to different types of cancer tissue. We demonstrate that the method generates realistic cancer gene expression data.
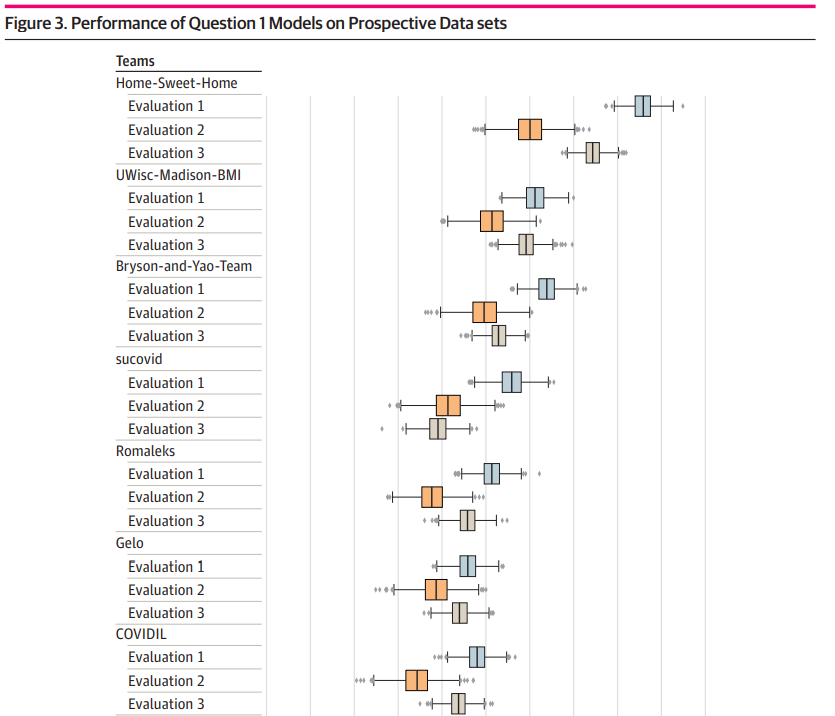
Yan, Yao, Thomas Schaffter, Timothy Bergquist, Thomas Yu, Justin Prosser, Zafer Aydin, Amhar Jabeer, et al.
JAMA Network Open 4, no. 10 (11 October 2021): e2124946.
This paper looked at the effectiveness of AI models at diagnosing Covid-19 from Electronic Health Records of patients in real time at the height of the Covid pandemic. Our model was ranked 3rd out of 90 teams.
Precision Medicine
Bentham, Robert B., Kevin Bryson, and Gyorgy Szabadkai.
Nucleic Acids Research 45, no. 15 (6 September 2017): 8712–30.
MCbiclust is a novel method that can extract patterns from massive transcriptomics data collections. We show that it can effectively stratify patients based on their intrinsic gene expression signatures.
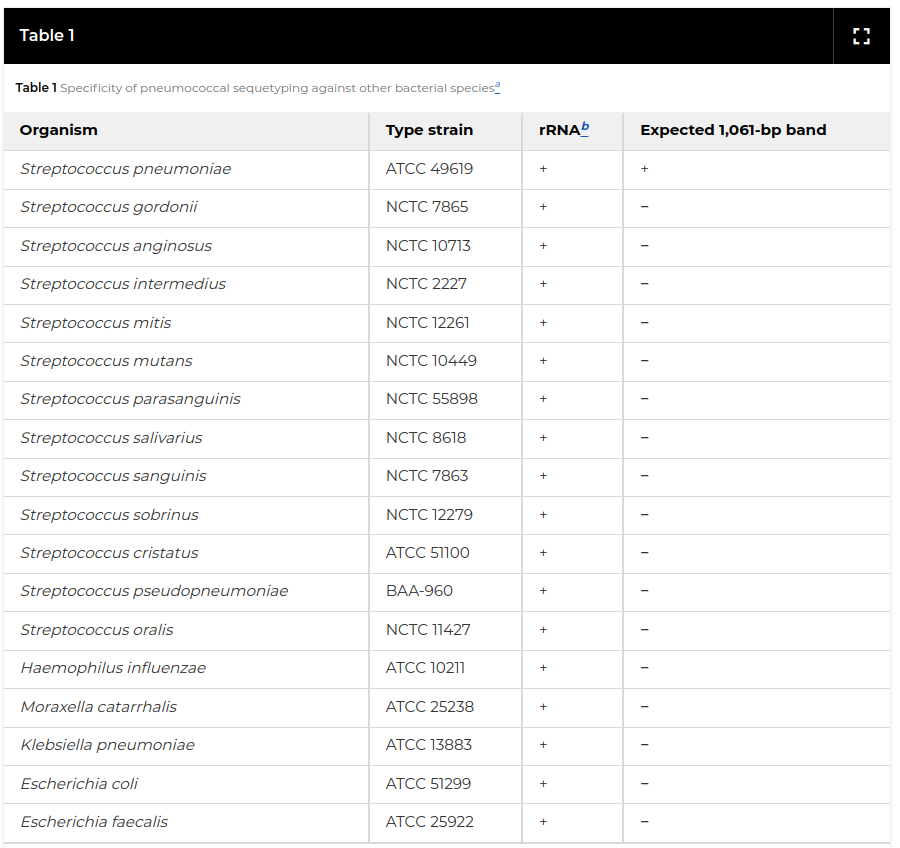
Leung, Marcus H., Kevin Bryson, Kathrin Freystatter, Bruno Pichon, Giles Edwards, Bambos M. Charalambous, and Stephen H. Gillespie.
Journal of Clinical Microbiology 50, no. 7 (21 December 2020): 2419–27.
A large-scale genomic analsis, based on decision trees, using PrimerFinder was used to identify the most informative PCR primer pairs for identifying different serotypes of streptococcus pneumoniae. This was experimentally validated in the lab and provides a cheap and easy way to monitor the spread of different serotypes across Tanzania and other countries.
Malki, K., E. Koritskaya, F. Harris, K. Bryson, M. Herbster, and M. G. Tosto.
Translational Psychiatry 6, no. 6 (June 2016): e839–e839.
This applies machine learning to epigenetic data from identical twins where one has major depressive disorder and the other does not. This identifies biological networks that potentially involved in the diseased mechanism.
Medlar, Alan, Dorota Głowacka, Horia Stanescu, Kevin Bryson, and Robert Kleta.
Bioinformatics 29, no. 4 (15 February 2013): 413–19.
Human pedigree analysis is often used to identify regions of the genome that are invovled in rare diseases. The analysis of large human pedigrees is incredibly computational; particularly consanguineous ones which often results in rare recessive diseases showing themselves. In this paper we develop an parallel MCMC genetic linkage algorithm that uses multicore CPUs and GPUs, resulting in a magnitute improvement in speed over other popular methods at the time.
Menegollo, Michela, Robert B. Bentham, Tiago Henriques, Seow Q. Ng, Ziyu Ren, Clarinde Esculier, Sia Agarwal, et al.
Cancer Research 84, no. 17 (4 September 2024): 2911–25.
Application of our MCbiclust software to breast cancer samples and experimental validation using metabolomics.
Patel, Z, Patel, ZI, Robinson, J, Robertson, DP, Stein, JE, Chenery, P, Latham, K, Evseeva, I, Bryson, K, and Marsh, SGE.
25th British Society for Histocompatibility and Immunogenetics Conference
A new probabilistic matching algorithm was developed which was used by the Anthony Nolan Trust for bone marrow transplant matching.
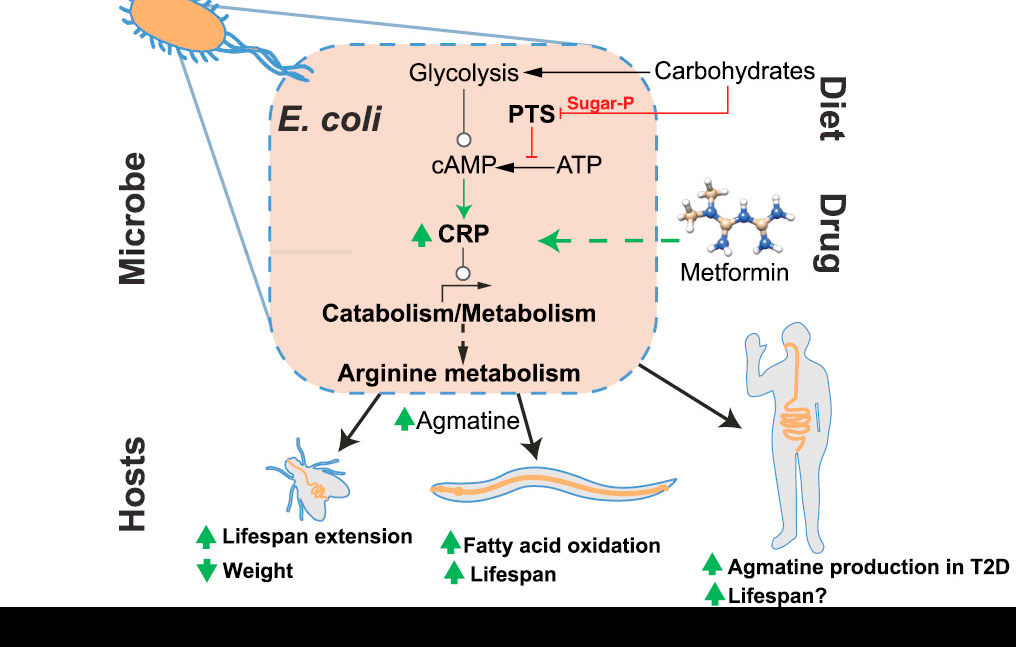
Pryor, Rosina, Povilas Norvaisas, Georgios Marinos, Lena Best, Louise B. Thingholm, Leonor M. Quintaneiro, Wouter De Haes, Daniela Esser, Silvio Waschina, and Celia Lujan.
Cell 178, no. 6 (2019): 1299–1312.
This study looked at how gut bacteria can alter the affect of drugs and nutrients on a host organism. It used the worm host C. elegans and the gut bacteria E. coli for high-throughout multi-omics screening. Data analysis of this four-way screen combined with metabolic modelling showed that microbes integrate cues from metformin and the diet through the phosphotransferase signaling pathway that converges on the transcriptional regulator Crp.
Scott, Timothy A., Leonor M. Quintaneiro, Povilas Norvaisas, Prudence P. Lui, Matthew P. Wilson, Kit-Yi Leung, Lucia Herrera-Dominguez, Sonia Sudiwala, Alberto Pessia, and Peter T. Clayton.
Cell 169, no. 3 (2017): 442–56.
Fluoropyrimidines are the first-line treatment for colorectal cancer, but their efficacy is highly variable between patients. We queried whether gut microbes, a known source of inter-individual variability, impacted drug efficacy. Integrating multi-omics data for our host/microbe system, our findings highlight the potential therapeutic power of manipulating intestinal microbiota to ensure host metabolic health and treat disease.
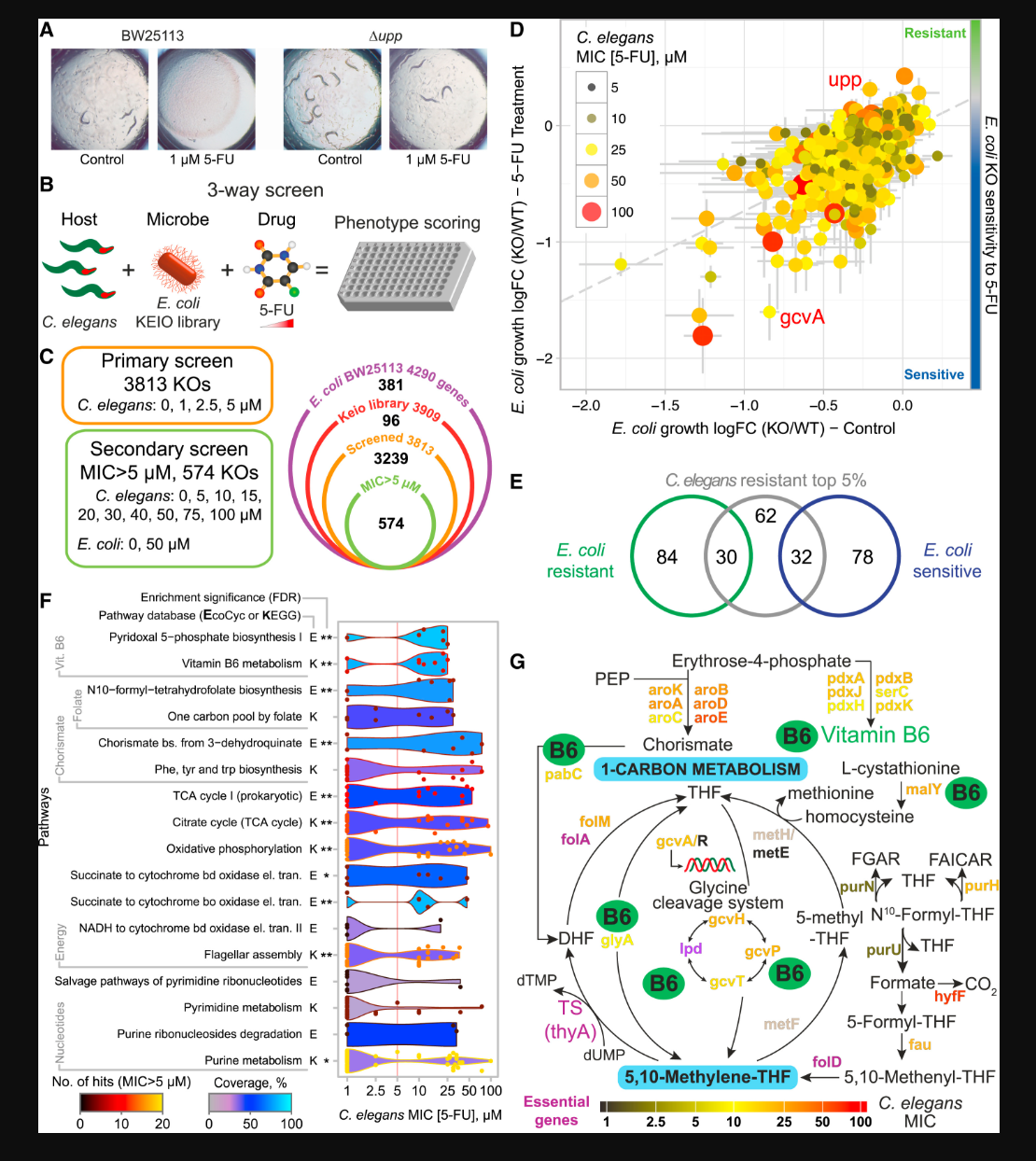
Yan, Yao, Thomas Schaffter, Timothy Bergquist, Thomas Yu, Justin Prosser, Zafer Aydin, Amhar Jabeer, et al.
JAMA Network Open 4, no. 10 (11 October 2021): e2124946.
This paper looked at the effectiveness of AI models at diagnosing Covid-19 from Electronic Health Records of patients in real time at the height of the Covid pandemic. Our model was ranked 3rd out of 90 teams.
Genomics / Genetics
Bryson, K., V. Loux, R. Bossy, P. Nicolas, S. Chaillou, M. van de Guchte, S. Penaud, et al.
Nucleic Acids Research 34, no. 12 (1 June 2006): 3533–45.
This system took the abstract ideas of the GeneWeaver multi-agent system and deployed it for genome annotation at INRA in France. A system was developed that allowed different labs at INRA to collaboratively annotate related species of lactobacillus (lactis, debrukei and sakei). Each lab had control over its own annotation processes while the systems collaborated on sharing annotation when relevant.
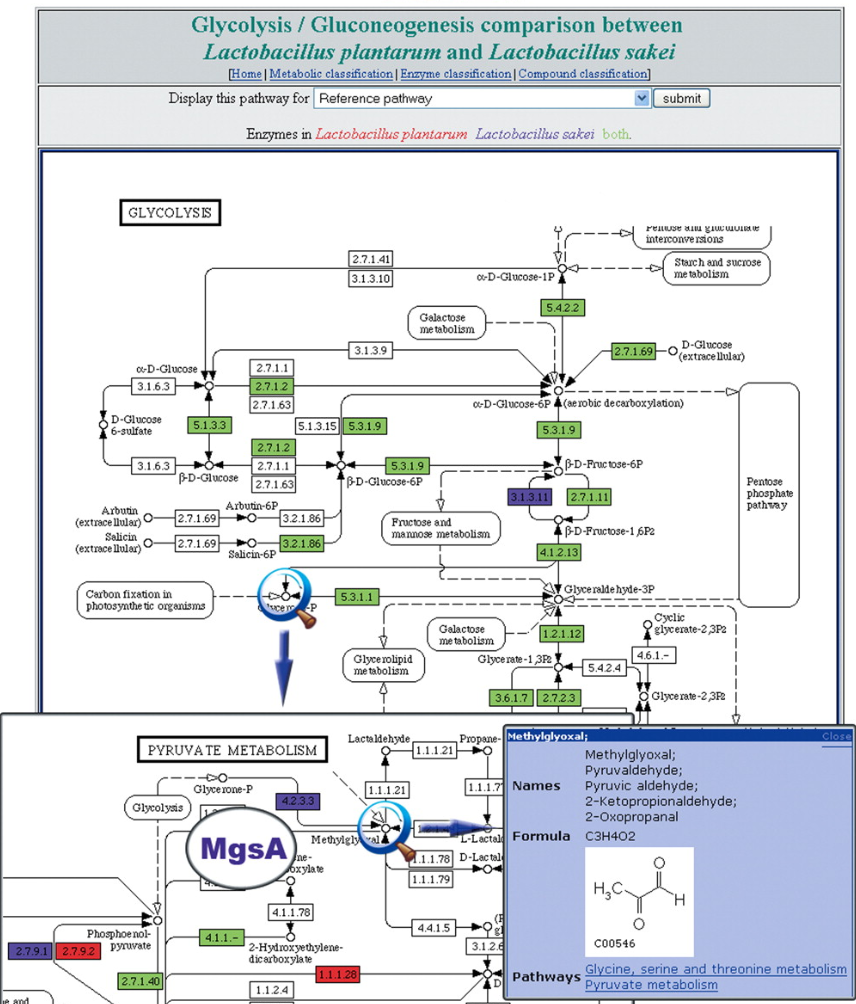
Bryson, K., M. Luck, M. Joy, and D. T. Jones.
Seminal paper outlining the GeneWeaver multi-agent system (MAS) for cooperative genome annotation.
Guchte, M. van de, S. Penaud, C. Grimaldi, V. Barbe, K. Bryson, P. Nicolas, C. Robert, et al.
Proceedings of the National Academy of Sciences 103, no. 24 (13 June 2006): 9274–79.
Thousands of years ago, humans accidentally introduced Lactobacillus Bulgaricus to milk where it produced yogurt. This paper used the AGMIAL genome annotation system to reveal the novel finding that this bacteria has been rapidly evolving over these thousands of years to move from its original plant host to the richer environment of milk, for instance, loosing genes for cellulose processing amongst other functions.
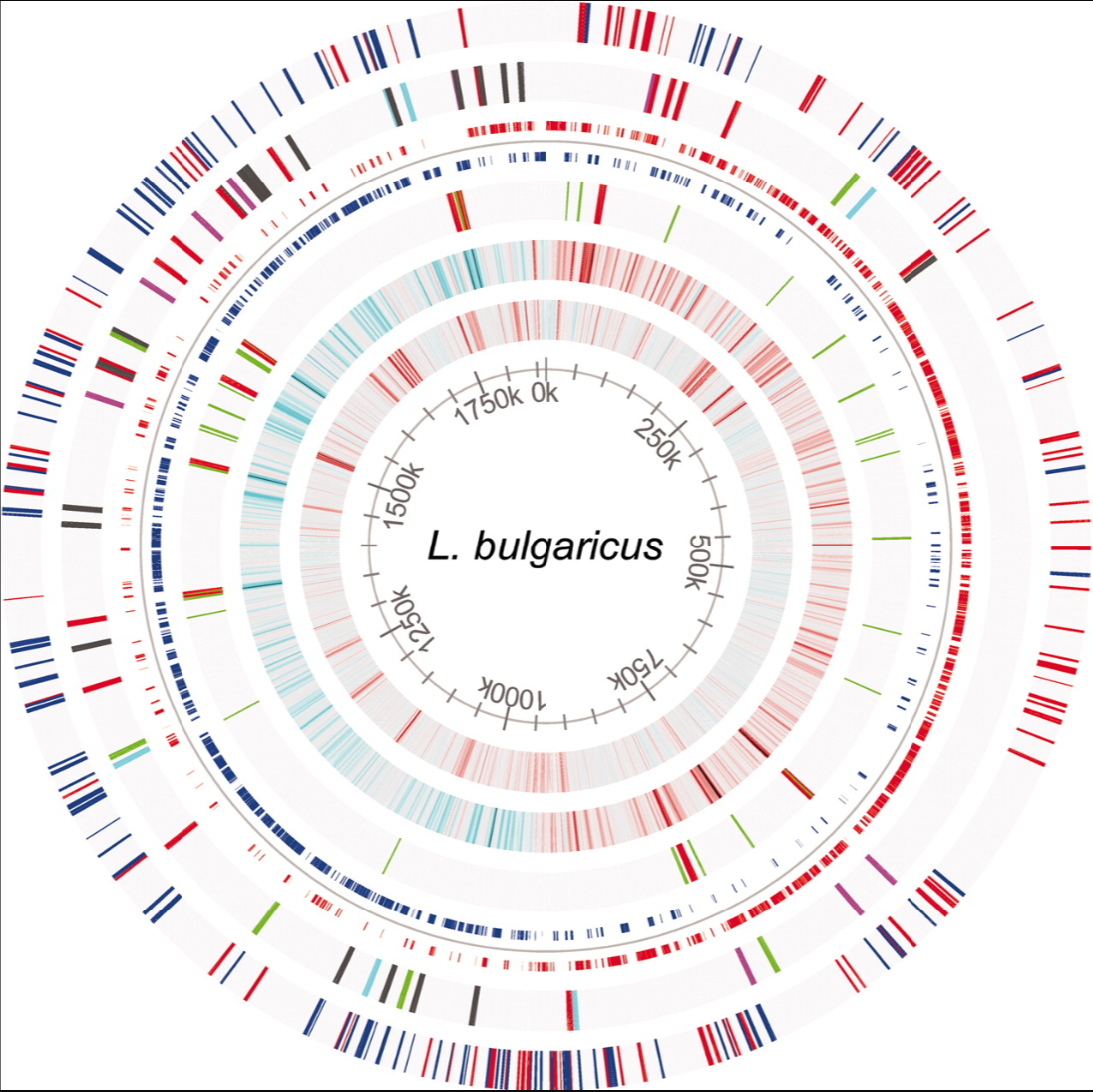
Leung, Marcus H., Kevin Bryson, Kathrin Freystatter, Bruno Pichon, Giles Edwards, Bambos M. Charalambous, and Stephen H. Gillespie.
Journal of Clinical Microbiology 50, no. 7 (21 December 2020): 2419–27.
A large-scale genomic analsis, based on decision trees, using PrimerFinder was used to identify the most informative PCR primer pairs for identifying different serotypes of streptococcus pneumoniae. This was experimentally validated in the lab and provides a cheap and easy way to monitor the spread of different serotypes across Tanzania and other countries.
Malki, K., E. Koritskaya, F. Harris, K. Bryson, M. Herbster, and M. G. Tosto.
Translational Psychiatry 6, no. 6 (June 2016): e839–e839.
This applies machine learning to epigenetic data from identical twins where one has major depressive disorder and the other does not. This identifies biological networks that potentially involved in the diseased mechanism.
Medlar, Alan, Dorota Głowacka, Horia Stanescu, Kevin Bryson, and Robert Kleta.
Bioinformatics 29, no. 4 (15 February 2013): 413–19.
Human pedigree analysis is often used to identify regions of the genome that are invovled in rare diseases. The analysis of large human pedigrees is incredibly computational; particularly consanguineous ones which often results in rare recessive diseases showing themselves. In this paper we develop an parallel MCMC genetic linkage algorithm that uses multicore CPUs and GPUs, resulting in a magnitute improvement in speed over other popular methods at the time.
Patel, Z, Patel, ZI, Robinson, J, Robertson, DP, Stein, JE, Chenery, P, Latham, K, Evseeva, I, Bryson, K, and Marsh, SGE.
25th British Society for Histocompatibility and Immunogenetics Conference
A new probabilistic matching algorithm was developed which was used by the Anthony Nolan Trust for bone marrow transplant matching.
Pryor, Rosina, Povilas Norvaisas, Georgios Marinos, Lena Best, Louise B. Thingholm, Leonor M. Quintaneiro, Wouter De Haes, Daniela Esser, Silvio Waschina, and Celia Lujan.
Cell 178, no. 6 (2019): 1299–1312.
This study looked at how gut bacteria can alter the affect of drugs and nutrients on a host organism. It used the worm host C. elegans and the gut bacteria E. coli for high-throughout multi-omics screening. Data analysis of this four-way screen combined with metabolic modelling showed that microbes integrate cues from metformin and the diet through the phosphotransferase signaling pathway that converges on the transcriptional regulator Crp.
Scott, Timothy A., Leonor M. Quintaneiro, Povilas Norvaisas, Prudence P. Lui, Matthew P. Wilson, Kit-Yi Leung, Lucia Herrera-Dominguez, Sonia Sudiwala, Alberto Pessia, and Peter T. Clayton.
Cell 169, no. 3 (2017): 442–56.
Fluoropyrimidines are the first-line treatment for colorectal cancer, but their efficacy is highly variable between patients. We queried whether gut microbes, a known source of inter-individual variability, impacted drug efficacy. Integrating multi-omics data for our host/microbe system, our findings highlight the potential therapeutic power of manipulating intestinal microbiota to ensure host metabolic health and treat disease.
Transcriptomics
Bentham, Robert B., Kevin Bryson, and Gyorgy Szabadkai.
Nucleic Acids Research 45, no. 15 (6 September 2017): 8712–30.
MCbiclust is a novel method that can extract patterns from massive transcriptomics data collections. We show that it can effectively stratify patients based on their intrinsic gene expression signatures.
Menegollo, Michela, Robert B. Bentham, Tiago Henriques, Seow Q. Ng, Ziyu Ren, Clarinde Esculier, Sia Agarwal, et al.
Cancer Research 84, no. 17 (4 September 2024): 2911–25.
Application of our MCbiclust software to breast cancer samples and experimental validation using metabolomics.
Pryor, Rosina, Povilas Norvaisas, Georgios Marinos, Lena Best, Louise B. Thingholm, Leonor M. Quintaneiro, Wouter De Haes, Daniela Esser, Silvio Waschina, and Celia Lujan.
Cell 178, no. 6 (2019): 1299–1312.
This study looked at how gut bacteria can alter the affect of drugs and nutrients on a host organism. It used the worm host C. elegans and the gut bacteria E. coli for high-throughout multi-omics screening. Data analysis of this four-way screen combined with metabolic modelling showed that microbes integrate cues from metformin and the diet through the phosphotransferase signaling pathway that converges on the transcriptional regulator Crp.
Scott, Timothy A., Leonor M. Quintaneiro, Povilas Norvaisas, Prudence P. Lui, Matthew P. Wilson, Kit-Yi Leung, Lucia Herrera-Dominguez, Sonia Sudiwala, Alberto Pessia, and Peter T. Clayton.
Cell 169, no. 3 (2017): 442–56.
Fluoropyrimidines are the first-line treatment for colorectal cancer, but their efficacy is highly variable between patients. We queried whether gut microbes, a known source of inter-individual variability, impacted drug efficacy. Integrating multi-omics data for our host/microbe system, our findings highlight the potential therapeutic power of manipulating intestinal microbiota to ensure host metabolic health and treat disease.
Viñas, Ramon, Helena Andrés-Terré, Pietro Liò, and Kevin Bryson.
Bioinformatics 38, no. 3 (12 January 2022): 730–37.
We introduce the first deep learning GAN method with word embeddings to simulate gene expression data for a wide range of biological systems from bacteria to different types of cancer tissue. We demonstrate that the method generates realistic cancer gene expression data.
Proteomics
McGuffin, Liam J., Kevin Bryson, and David T. Jones.
Bioinformatics 16, no. 4 (1 April 2000): 404–5.
One of the most successful neural network architectures for predicting ab-initio secondary structure from protein sequences.
Pryor, Rosina, Povilas Norvaisas, Georgios Marinos, Lena Best, Louise B. Thingholm, Leonor M. Quintaneiro, Wouter De Haes, Daniela Esser, Silvio Waschina, and Celia Lujan.
Cell 178, no. 6 (2019): 1299–1312.
This study looked at how gut bacteria can alter the affect of drugs and nutrients on a host organism. It used the worm host C. elegans and the gut bacteria E. coli for high-throughout multi-omics screening. Data analysis of this four-way screen combined with metabolic modelling showed that microbes integrate cues from metformin and the diet through the phosphotransferase signaling pathway that converges on the transcriptional regulator Crp.
Radivojac, Predrag, Wyatt T. Clark, Tal Ronnen Oron, Alexandra M. Schnoes, Tobias Wittkop, Artem Sokolov, Kiley Graim, et al.
Nature Methods 10, no. 3 (March 2013): 221–27.
A competition involving prediction Gene Ontology protein function prediction for protein sequences. The method of our research group at UCL was ranked first in the competition.
Scott, Timothy A., Leonor M. Quintaneiro, Povilas Norvaisas, Prudence P. Lui, Matthew P. Wilson, Kit-Yi Leung, Lucia Herrera-Dominguez, Sonia Sudiwala, Alberto Pessia, and Peter T. Clayton.
Cell 169, no. 3 (2017): 442–56.
Fluoropyrimidines are the first-line treatment for colorectal cancer, but their efficacy is highly variable between patients. We queried whether gut microbes, a known source of inter-individual variability, impacted drug efficacy. Integrating multi-omics data for our host/microbe system, our findings highlight the potential therapeutic power of manipulating intestinal microbiota to ensure host metabolic health and treat disease.
Metabolomics
Bryson, K., V. Loux, R. Bossy, P. Nicolas, S. Chaillou, M. van de Guchte, S. Penaud, et al.
Nucleic Acids Research 34, no. 12 (1 June 2006): 3533–45.
This system took the abstract ideas of the GeneWeaver multi-agent system and deployed it for genome annotation at INRA in France. A system was developed that allowed different labs at INRA to collaboratively annotate related species of lactobacillus (lactis, debrukei and sakei). Each lab had control over its own annotation processes while the systems collaborated on sharing annotation when relevant.
McBride, Ross, Joe Wandy, Stefan Weidt, Simon Rogers, Vinny Davies, Rónán Daly, and Kevin Bryson.
Bioinformatics 39, no. 7 (1 July 2023): btad406.
Presented a number of novel algorithms for mass spectrometry instruments that showed a significant improvement over previous ones.
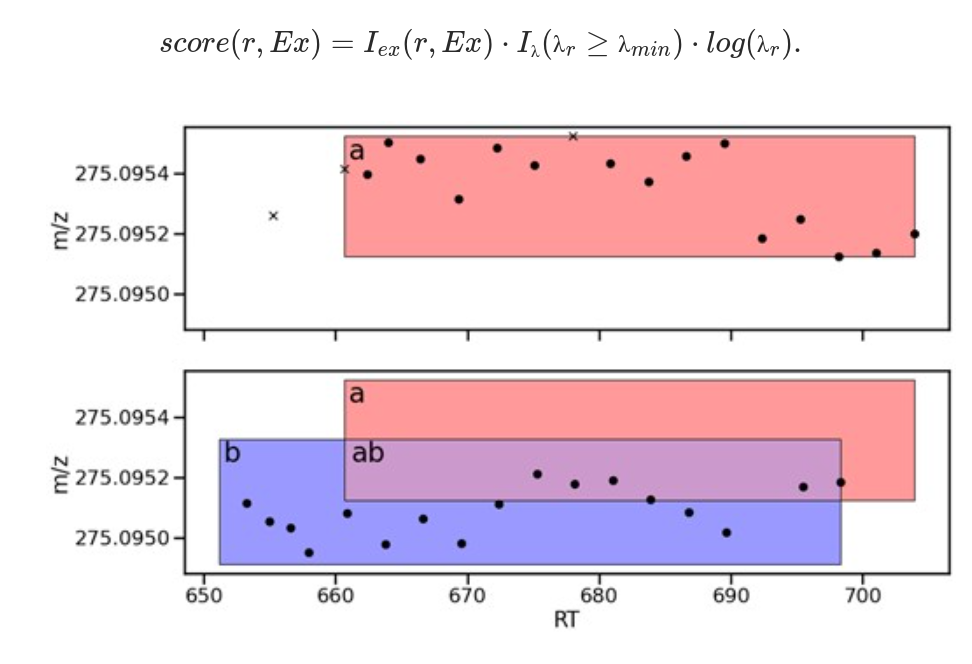
Pryor, Rosina, Povilas Norvaisas, Georgios Marinos, Lena Best, Louise B. Thingholm, Leonor M. Quintaneiro, Wouter De Haes, Daniela Esser, Silvio Waschina, and Celia Lujan.
Cell 178, no. 6 (2019): 1299–1312.
This study looked at how gut bacteria can alter the affect of drugs and nutrients on a host organism. It used the worm host C. elegans and the gut bacteria E. coli for high-throughout multi-omics screening. Data analysis of this four-way screen combined with metabolic modelling showed that microbes integrate cues from metformin and the diet through the phosphotransferase signaling pathway that converges on the transcriptional regulator Crp.
Scott, Timothy A., Leonor M. Quintaneiro, Povilas Norvaisas, Prudence P. Lui, Matthew P. Wilson, Kit-Yi Leung, Lucia Herrera-Dominguez, Sonia Sudiwala, Alberto Pessia, and Peter T. Clayton.
Cell 169, no. 3 (2017): 442–56.
Fluoropyrimidines are the first-line treatment for colorectal cancer, but their efficacy is highly variable between patients. We queried whether gut microbes, a known source of inter-individual variability, impacted drug efficacy. Integrating multi-omics data for our host/microbe system, our findings highlight the potential therapeutic power of manipulating intestinal microbiota to ensure host metabolic health and treat disease.
Wandy, Joe, Ross McBride, Simon Rogers, Nikolaos Terzis, Stefan Weidt, Justin J. J. van der Hooft, Kevin Bryson, Rónán Daly, and Vinny Davies.
Frontiers in Molecular Biosciences 10 (7 March 2023).
A significant finding of this paper is the application of the ViMMS ‘digital twin’ to do optimization of mass spectrometry instruments that would be expensive, difficult or impossible to do on real instruments.
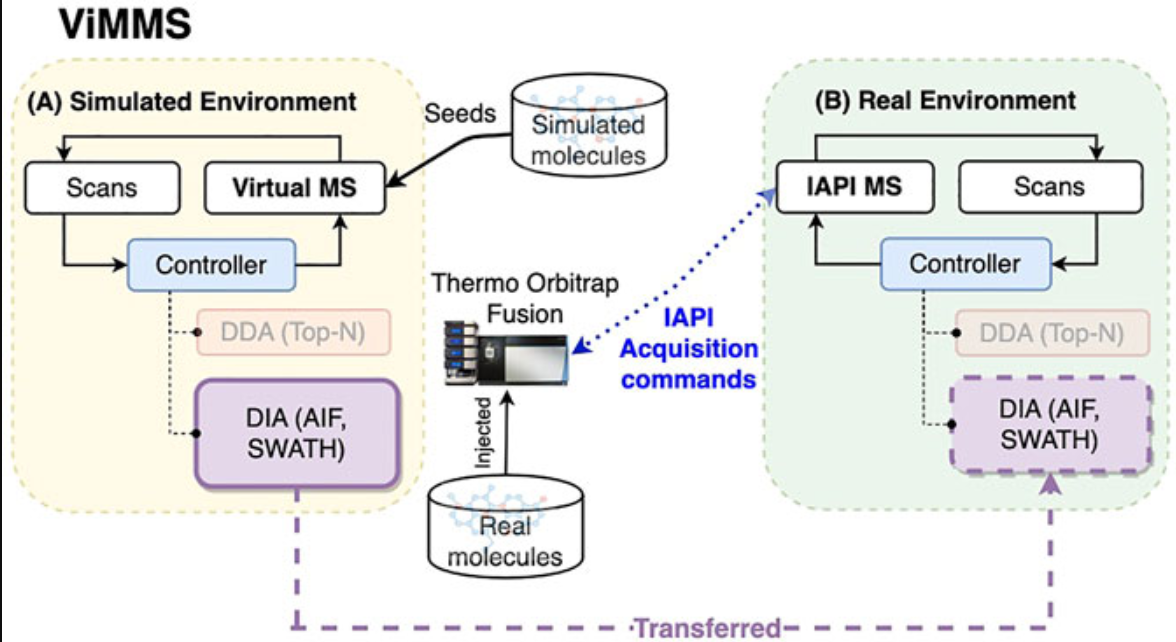
Systems Biology Integration
Bryson, K., V. Loux, R. Bossy, P. Nicolas, S. Chaillou, M. van de Guchte, S. Penaud, et al.
Nucleic Acids Research 34, no. 12 (1 June 2006): 3533–45.
This system took the abstract ideas of the GeneWeaver multi-agent system and deployed it for genome annotation at INRA in France. A system was developed that allowed different labs at INRA to collaboratively annotate related species of lactobacillus (lactis, debrukei and sakei). Each lab had control over its own annotation processes while the systems collaborated on sharing annotation when relevant.
Bryson, K., M. Luck, M. Joy, and D. T. Jones.
Seminal paper outlining the GeneWeaver multi-agent system (MAS) for cooperative genome annotation.
Pryor, Rosina, Povilas Norvaisas, Georgios Marinos, Lena Best, Louise B. Thingholm, Leonor M. Quintaneiro, Wouter De Haes, Daniela Esser, Silvio Waschina, and Celia Lujan.
Cell 178, no. 6 (2019): 1299–1312.
This study looked at how gut bacteria can alter the affect of drugs and nutrients on a host organism. It used the worm host C. elegans and the gut bacteria E. coli for high-throughout multi-omics screening. Data analysis of this four-way screen combined with metabolic modelling showed that microbes integrate cues from metformin and the diet through the phosphotransferase signaling pathway that converges on the transcriptional regulator Crp.
Scott, Timothy A., Leonor M. Quintaneiro, Povilas Norvaisas, Prudence P. Lui, Matthew P. Wilson, Kit-Yi Leung, Lucia Herrera-Dominguez, Sonia Sudiwala, Alberto Pessia, and Peter T. Clayton.
Cell 169, no. 3 (2017): 442–56.
Fluoropyrimidines are the first-line treatment for colorectal cancer, but their efficacy is highly variable between patients. We queried whether gut microbes, a known source of inter-individual variability, impacted drug efficacy. Integrating multi-omics data for our host/microbe system, our findings highlight the potential therapeutic power of manipulating intestinal microbiota to ensure host metabolic health and treat disease.
